Overview
EU IVDR Compliance Services Overview
European In Vitro Diagnostic Regulation (EU IVDR) is a new Regulatory basis for placing IVDs in the European market. The EU IVDR Regulation is set to replace the EU’s current Directive on in vitro diagnostic medical devices (EU 98/79/EC). As a European regulation, it will be effective in all the EU Member States and the European Free Trade Association (EFTA) states without any need to be transferred into the law of respective States. As a proven Regulatory partner, Freyr provides EU IVDR compliance services and IVDR consulting services to manufacturers for compliant In Vitro Diagnostic Regulations and CE marking.
Book a meeting with our EU IVDR experts
The IVDR Regulation - Transition Timeline
Mandated to be effective from May 26, 2022, the new EU IVDR is considered as a significant change to the Regulatory oversight of IVDs. Overview of the IVDR transition is as below –
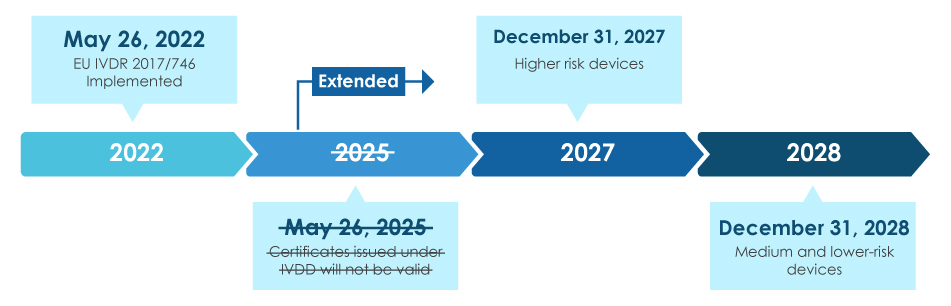
Once fully implemented, the IVDR compliance rules ensure that almost all IVDs entering the European market are subject to the IVDR Notified Body (NB) review and the CE marking certification as a part of the IVD Approval Process. In this scenario, apart from aligning with the updated IVDR classification, there is an immediate requirement for manufacturers to effectively review the key technical documentation for successful IVD registration and CE marking. The IVDR requirements vary with the risk class of the IVD. In general, for the IVD Certification, manufacturers must perform:
- Technical file reviews as per the IVDR regulation (Regulation EU 2017/746)
- Performance Evaluation Report (PER) preparation for all the IVDs
- Post-market Performance Follow-up (PMPF) as per the Annex XIII Part B of the IVDR
- Vigilance Reporting as per the Article 82 of the IVDR
Freyr supports clients in performing a systematic review of the scientific literature and aid in the planning and preparation of a PER for In Vitro Diagnostic Regulation compliance. Freyr has dedicated experts who provide IVDR consulting services and Post-market Surveillance (PMS) support, which is an integral part of the manufacturer’s quality management system along with Post-market Performance Follow-up (PMPF).
Freyr Expertise and Advantages
Expertise
Freyr Expertise
- Transition plan for the IVDR compliance
- Technical review and gap analysis of the IVDR requirements for GSPR (General Safety and Performance Requirements)
- Support the compilation of the technical file as per the IVDR requirements
- Scientific validity reports based on literature and/or in-house data
- Clinical performance reports based on literature and/or in-house data
- Clinical evidence or Performance Evaluation reports
- Post-market Performance Follow up (PMPF) protocols and reports
- Post-market Surveillance (PMS) protocols and reports
- Writing/revising other documents such as package insert/IFU (Instructions for Use), Quick Reference Instructions (QRI), Operation/User Manual etc.
Advantages
Freyr Advantages
- Assured IVDR compliance, IVD registration, and CE marking
- Strong Regulatory understanding and expertise in the EU IVDR key impact areas
- Strong project management driven delivery model to ensure schedule adherence
- In-house NB Experts (review of the report by the NB interactive reviewers)
- Focused teams with cross expertise on specific impact areas and device categories
- Cross-functional inputs from medical device experts to comply with requirements
- Full scope of service across compliance, review and planning
- Strong expertise in maintaining consistency in deliverables (Time and Quality)
For end-to-end regulatory support on EU IVDR, reach out to Freyr