Turkey Medical Device Registration Overview
The Turkish medical device market has seen significant and consistent growth over the past decade. From 2021onwards, Turkey Medical Device Registration Requires to abide by the EU EU Medical Devices Regulation (MDR) 2017/745 and In Vitro Diagnostics Medical Devices Regulation (IVDR) 2017/746. This enhanced international trade leading several global companies to launch their medical devices in the country.
Regulatory Authority: Turkish Medicines and Medical Devices Agency (TITCK)
Regulation: Medical Device Regulations (MDR) 2017/745, In Vitro Diagnostic Devices Regulations 2017/746
Regulatory Pathway: CE marking is mandatory followed by Registration/Notification in the Product Tracking System (UTS)
Turkey Local Authorized Representative: European Authorized Representative (EAR) for foreign (non-EU/non-Turkey) manufacturers
QMS Requirement: ISO 13485:2016
Assessment of Technical Data: Notified Body for CE marking
Validity of License: Unlimited
Submission Format: Paper
Translation: Translated Documents in Turkish
Device Classification
Turkey follows the same medical device classification as given in the EU MDR and IVDR. Determining the device classification can be challenging and hence having backed by an experienced regulatory consultant is quite crucial here.
Medical Device Classes -
|
Class |
Risk |
|
Class I |
Low |
|
Class IIa |
Moderate |
|
Class IIb |
Moderate to High |
|
Class III |
High |
In Vitro Diagnostic Device Classes -
|
Class |
Risk |
|
Class A |
Low |
|
Class B |
Moderate |
|
Class C |
Moderate to High |
|
Class D |
High |
Turkey Local Authorized Representative
Now, due to the custom union agreement, the EU manufacturers do not require to appoint a local authorised representative to place their devices in the market.
Other foreign manufacturers are required to appoint a European Authorised Representative (EAR) to place the devices in the Turkey market.
Medical Device Registration
CE marking is a conformity that is required by the manufacturers to place their device in the Turkey market. CE marking is issued through a conformity assessment carried out by the notified body. Now, Turkey is authorised to appoint notified bodies in accordance with EU MDR and IVDR.
The companies are required to register in Central Registration System (MERSIS) registration and register the device in the Product Tracking System (UTS).
Process flow
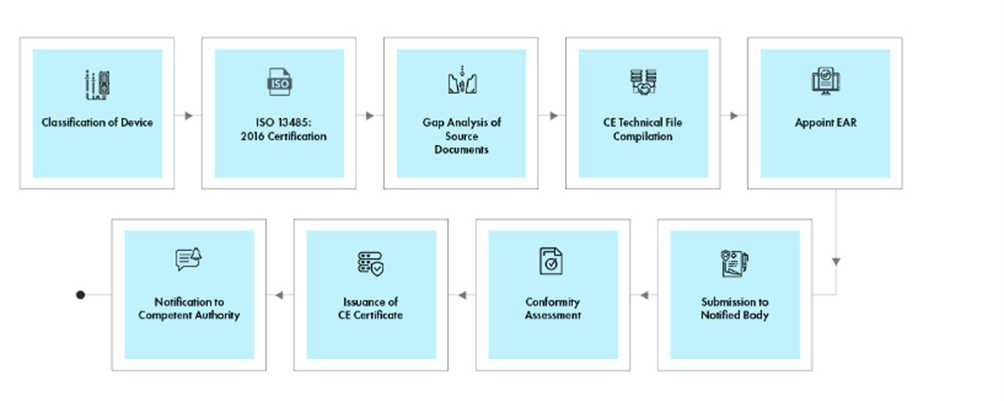
Post-approval Device Lifecycle Management
Freyr supports foreign manufacturers in end-to-end Medical Device lifecycle management, including post-approval activities, such as:
- Post approval change management - modifications to existing medical device approvals such as the addition of new variants, accessories; addition of new indications of use among others
- Maintenance of ISO 13485:2016 and CE certification
- Renewal of licenses
- Liaising between Notified body and the manufacturer
With various authorization bodies involved, foreign manufacturers need to comply with multiple sets of regulations in each individual process for device approvals. Obtaining a CE marking and further adhering to state-wise regulations needs extensive Regulatory knowledge. At times, without a proven Regulatory partner, navigating through all the device requirements might be challenging for market entrants. To assist manufacturers, Freyr provides end-to-end Regulatory services to expedite approvals for medical devices.
Expertise
Freyr Expertise
- European Medical Device Classification
- European Authorized Representative (EAR) support
- Turkey Device Registration and Product Notification
- ISO 14971:2019 Risk Management consultation
- ISO 13485:2016 Compliance
- CE technical file/design dossier review, compilation, and submission
- EU MDR Transition Support
- EU IVDR Transition Support
- Clinical Evaluation Reports (CER) for Medical Devices
- Performance Evaluation Reports (PER) for In Vitro Diagnostic Devices
- Notification/Registration of Medical Devices via Online Registration System
- Medical Device Regulatory strategy report
- Testing support- biocompatibility, electrical safety, mechanical and performance
- Labeling Compliance support
- GMP support
- Post-market Surveillance support