UK Medical Device Registration Overview
Post-Brexit, the UK is still altering and adding its regulations for medical devices. The regulations to be followed in the country are bifurcated geographically – Great Britain (GB) and Northern Ireland (NI). Medicines and Healthcare Products Regulatory Agency (MHRA) is the regulatory authority that looks after medical device. Northern Ireland is required to comply with European Union Medical Devices Regulations (EU MDR) 2017/745 and In Vitro Diagnostic Devices Regulations (IVDR) 2017/746. Non-UK manufacturers must appoint a UK Responsible Person (UK RP) to help them comply with these regulations and ensure a successful UK Medical Device Registration process.
Regulatory Authority: Medicines and Healthcare Products Regulatory Agency (MHRA)
Regulation: Medical Devices Regulations (MDR) 2002*
Regulatory Pathway: CE marking followed by Notification
Authorized Representative: UK Responsible Person (UK RP) for Non-UK manufacturers
QMS Requirement: ISO 13485:2016
Assessment of Technical Data: UK Approved Bodies for UKCA marking
Valid Markings: GB – UKCA or CE & NI – CE or CE + UKNI
Submission Format: Paper
*Future Regulations for medical devices to apply from Jul 1, 2025
The UK Medical Device Classification
The UK Medical device classifications are based on the UK MDR 2002. Classifying the device is the very first step in the entire process of obtaining approval and launching it in the market.
Medical Device Classification
|
Class |
Risk |
|
Class I |
Low |
|
Class IIa |
Medium |
|
Class IIb |
Medium |
|
Class III |
High |
IVD Classification
- General IVD
- Self-testing IVDs
- IVDs considered under Annex II List A
- IVDs considered under Annex II List B
Our company specializes in medical device classification. Our Medical Device consultants has successfully completed device classification for over 50 companies to date in the UK.
United Kingdom Responsible Person (UKRP) Services
Non-UK manufacturers are now required to appoint UKRP mandatorily to place the products in the market.
Our Medical Device consultants has successfully registered and can now act as your UKRP. To get more details on our UKRP services, please visit – www.ukrpservices.com
Medical Device Registration
The manufacturers will now have to appoint a UK-approved body to obtain UKCA marking. Though CE marking is allowed, it is applicable only for a certain time period. The transition timelines for the same are mentioned below –
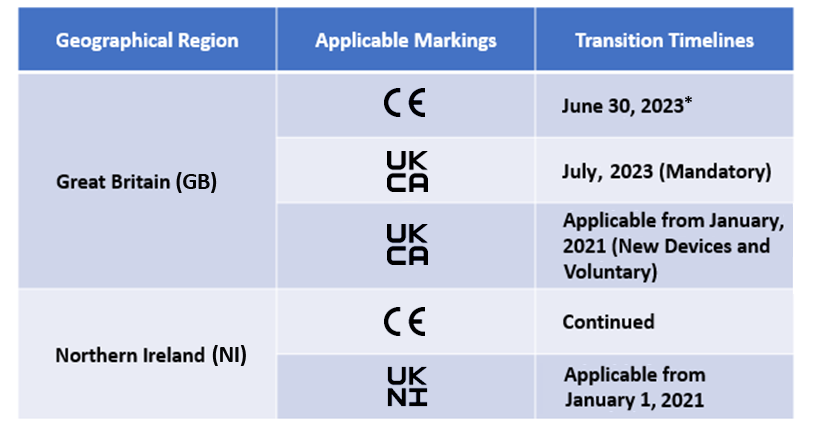
Our Medical Device consultants is currently supporting many manufacturers with the post-brexit transitions.
*The current transition timeline for CE marking under EU MDR/EU IVDR is recognised by UK MHRA and the timelines will differ as per the scope of the devices
Post-market Surveillance Requirements
Post-market surveillance requirements under UK MDR 2002 are quite stringent to ensure patient/user safety and effectiveness. Currently, the PMS activities include reporting of incidents/findings to the MHRA. The MHRA has released comprehensive guidance on the same.
Expertise
Freyr Expertise
- UK Medical Device Classification
- Regulatory Transition Support concerning post-brexit
- Regulatory Support for UK MHRA Notification
- United Kingdom Responsible Person (UKRP)
- Health Agency and Approved Body liaising and support