Overview
EU MDR Compliance Services Overview
The EU Medical Device Regulation (MDR) has come into effect from 26th May 2021, after the 3-year transition timeline and an additional one-year extension due to the COVID-19 pandemic. The devices which are being launched in the EU market now must comply with these regulations and shall be CE certified as per EU MDR by the Notified Bodies accredited under these regulations. The devices which have already been CE certified as per EU MDD however, have grace periods before they have to fully comply with the EU MDR requirements. During this grace period, the devices certified under both EU MDD and EU MDR will co-exist in the market with equal status and without being subjected to discrimination. Freyr offers unmatchable EU MDR Compliance services to help medical device companies meet the EU MDR requirements in a timely manner.
Book a meeting with our EU MDR experts
The MDR - Transition Timeline and New Device Classifications
The European Medical Device Regulation (MDR) will be fully effective in all the EU member states and the European Free Trade Association (EFTA) States from May 2021 and provides manufacturers a transition period of 4 years for complete EU MDR Certification.
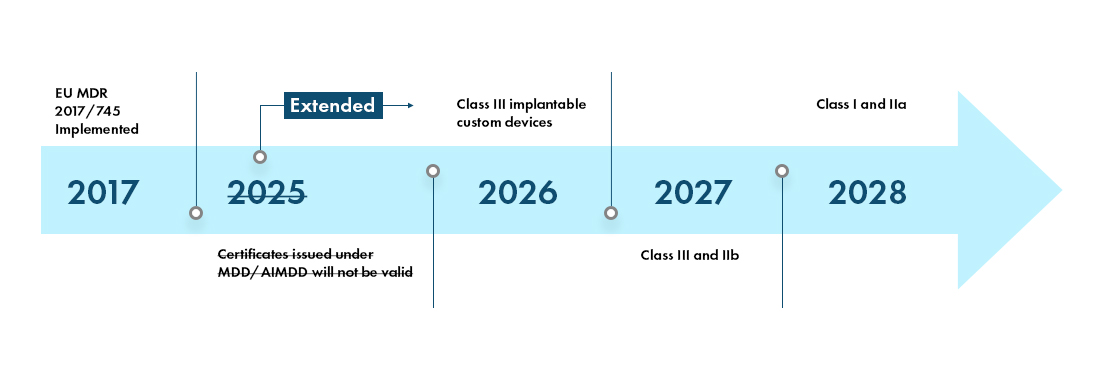
The new European Medical Device Regulation (MDR), as observed, have also brought changes to existing device classification system such as:
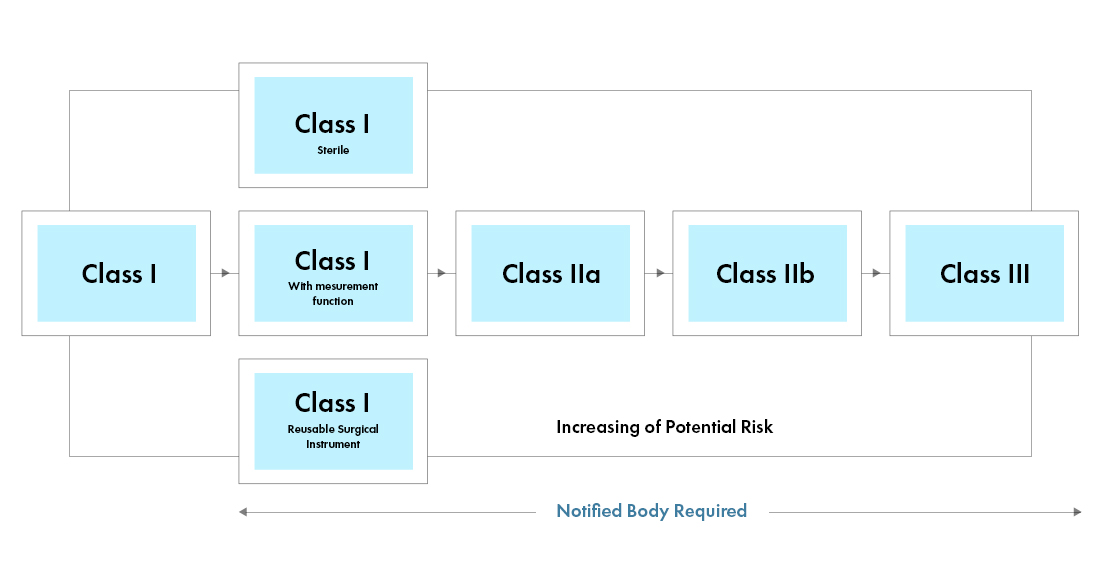
From identifying exact changes to be made to implementing them in real-time, manufacturers may have to navigate through array of challenges to comply with EU MDR requirements. Right from decoding the new structure, classifying a device accurately, to collate and submit all the data, a more detailed and cross-functional Regulatory approach will be required for manufacturers, to cope with the new European Medical Device Regulations. With a stringent gap analysis, Freyr assists clients with the status-quo and there by provides necessary Regulatory action needed for the transition and EU MDR Compliance.
Freyr Expertise and Advantages
Expertise
Freyr Expertise
- Developing a clear Medical Device Regulation (MDR) implementation strategy
- Understanding the new legislation, conducting Gap Analysis to current Quality Management Systems (QMS) and processes in place
- Developing a detailed plan with a cross-functional approach to determine aspects of quality system that will need modification in purview with the new EU Medical Device Regulation
- Forming multiple teams for analyzing product scope, classification, handling QMS etc., within organization, with a single point of contact in each team
- Allocation and planning of resources
- Considering your QMS interaction with other regulations and utilizing this opportunity to streamline processes, while allowing flexibility to incorporate future changes
- Analyzing the test data in place and check for any additional requirements that MDR puts in place
- Coordinating expectations and transition plan with your EU Notified Bodies
- Gap Analysis for existing Medical Devices from the EU MDD to the EU MDR Regulations
- End-to-end support to develop Clinical Evaluation Report (CER), including, literature search as per European Medical Device Regulation (EU MDR) guidelines
- End-to-end services for Post-market Surveillance Reports (PMSR), Periodic Safety Update Report (PSUR) and Summary of Safety and Clinical Performance (SSCP)
- Regulatory Resource Augmentation with both onshore and offshore deployment options
- European Authorized Representative (EAR) Services
- MDR Compliance and submission assistance to Notified Bodies
- Regulatory Intelligence covering importation process of different regulated markets
- QMS Compliance and mock audits
- Document Management System and tool for MDR companies
- Classification and reclassification of devices according to risk
- UDI implementation and consulting
- EU Medical Device Regulation Compliant Post-market Surveillance services
- Risk Management ISO 14971 consulting
- In-house and online training
- Person responsible for Regulatory compliance services and assistance
- Identification of MDR Notified Bodies
For end-to-end regulatory support on EU MDR, reach out to Freyr