Israel's medical device industry is experiencing sustained growth and innovation, making it a hub for cutting-edge healthcare technologies. Registering medical devices is crucial for companies entering this dynamic market. This overview explores key aspects of the Israel registration process, offering insights into the Regulatory framework and requirements for bringing innovative medical devices to the forefront of Israel's healthcare sector.
Regulatory Authority: The Medical Device Division of the Israeli Ministry of Health (AMAR).
Regulation: Medical Device/Equipment Law of 2012
Regulatory Pathway: Product Registration
Israel Local Authorized Representative: Israel Registration Holder (IRH)
QMS Requirement: ISO 13485
Assessment of Technical Data: The Medical Devices Department at the Ministry of Health
Validity of License: Five (05) years
Submission Format: Paper and Electronic
Translation: Translated Documents in Hebrew
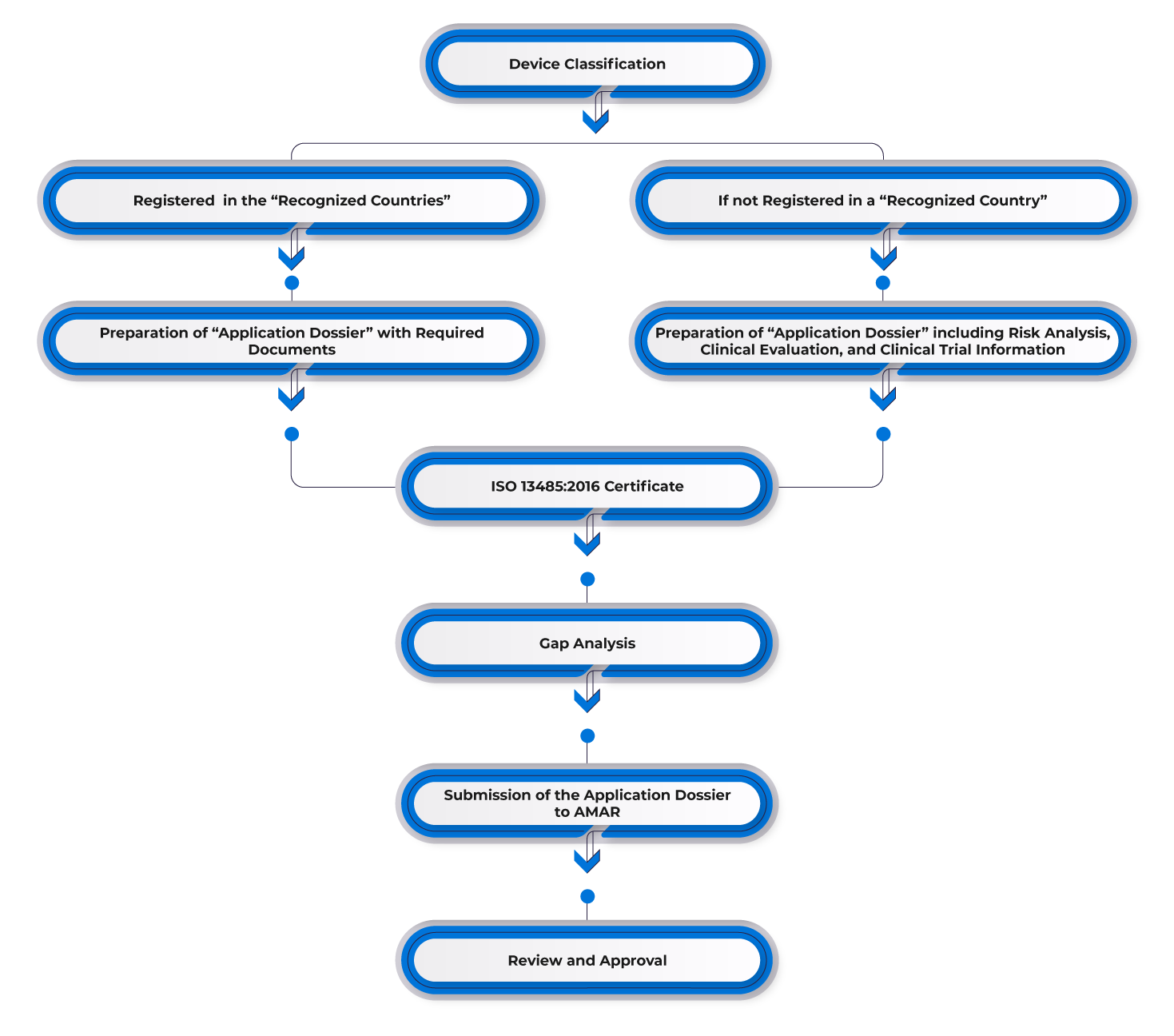
Device Classification
The Medical Equipment Law and Regulations for Medical Equipment Registration in Israel do not specify a risk classification system. Instead, Israel aligns its medical device classification with international standards, particularly those outlined by the Global Harmonization Task Force (GHTF) countries. Alternatively, the risk classification of a device in a recognized country is adopted for registration in Israel. This classification process typically considers the intended use, risk level, and other factors that may impact the safety and effectiveness of medical devices.
Medical Device Classes
|
Class |
Risk |
|
Class I |
Low |
|
Class II |
Low-Medium |
|
Class III |
High |
Changes Proposed to Registration Routes
Proposed modifications apply to Class I and Class II devices, while the registration system for Class III devices remains unchanged.
- Class I devices can be immediately registered through self-declaration.
- Class II devices, while declarations and technical documents are necessary, AMAR may expedite the process to fourteen (14) days for those deemed low-medium risk. This applies if the manufacturer holds two (02) authorizations from recognized countries and provides six (06) months of market data. Alternatively, for Class II devices with only the US FDA 510(k) clearance and six (06) months of the US market data, AMAR processing time is accelerated to sixty (60) days.
Israel Local Authorized Representative
Medical device firms based outside of Israel must designate an Israeli Registration Holder (IRH) to facilitate the registration of their products for sale within the country. The IRH acts as the manufacturer’s local representative and is tasked with liaising with the Ministry of Health to ensure adherence to local regulations. Additionally, an IRH is responsible for establishing and sustaining a business presence in Israel, as well as obtaining and upkeeping a valid business license.
Medical Device Registration
To register a medical device in Israel, manufacturers must secure prior approval in one of the reference markets such as the USA, Europe, Australia, Canada, or other major markets. Manufacturers with existing approvals in one of the reference countries can utilize this approval for the Israeli market and appoint an in-country representative. Subsequently, they need to submit the required documentation, including:
- FDA 510(k)/Premarket Approval Letter/CE.
- Certificate to Foreign Government (CFG)/Certificate of Free Sale (CFS).
- ISO 13485 or Another Recognized Good Manufacturing Practices (GMPs) Certification.
- Validation and Certification from the Standards Institute of Israel (If necessary).
Process Flow
Post-approval Device Lifecycle Management
Freyr extends comprehensive support to foreign manufacturers in managing the entire lifecycle of medical devices in Israel, including post-approval activities:
- Post-approval change management, addressing modifications to existing medical device approvals, such as the addition of new variants, accessories, and indications of use.
- Maintenance of ISO 13485:2016 and CE certification.
- Renewal of licenses.
- Acting as an intermediary between the Notified Body (NB) and the manufacturer.
Navigating the intricacies of authorization bodies and complying with multiple sets of regulations for device approvals can be challenging. Obtaining approvals from various GHTF countries and adhering to state-wise regulations require in-depth Regulatory knowledge. For market entrants facing these complexities without an established Regulatory partner, Freyr offers end-to-end Regulatory services, streamlining the approval process for medical devices in Israel.
Expertise
Freyr Expertise
- Israel Medical Device Classification.
- Israel Registration Holder (IRH).
- Israel Device Registration.
- ISO 14971:2019 Risk Management Consultation.
- ISO 13485:2016 Compliance.
- Design Dossier Review, Compilation, and Submission.
- Registration of Medical Devices via Online Registration System.
- Medical Device Regulatory Strategy Report.
- Testing Support- Biocompatibility, Electrical Safety, Mechanical and Performance.
- Labeling Compliance Support.
- GMP Support.
- Post-market Surveillance (PMS) Support.