Overview
Overview of The FDA eSTAR
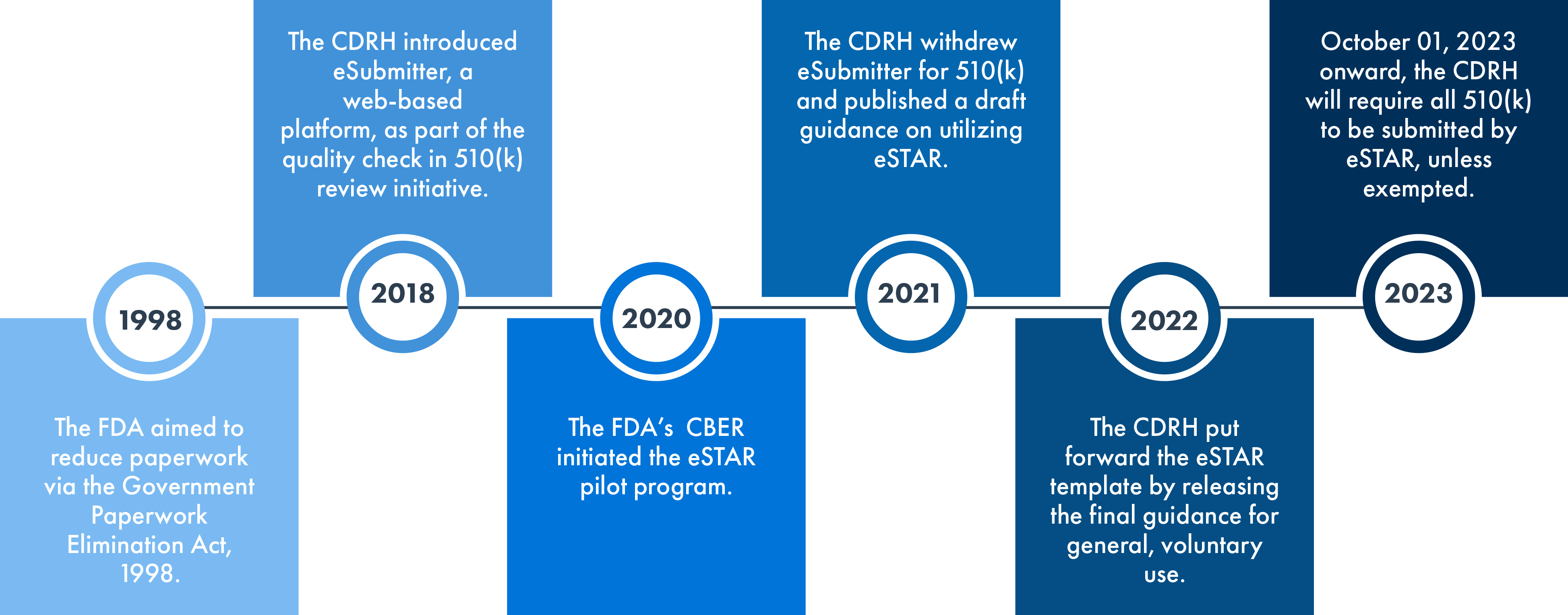
The FDA eSTAR initiative was formulated to enhance efficiency and uniformity in preparing and assessing the FDA 510(k) and De Novo submissions. Stemming from the initial eSubmitter approach, which involved the electronic submission of medical devices and IVD applications, the eSTAR program was established, drawing on the FDA’s prior experience. The FDA pioneered a different program called the Electronic Submission Template and Resource (eSTAR) Pilot Program. From October 01, 2023, 510(k) submissions (Traditional, Abbreviated, or Special 510[k]) will have to use the eSTAR program. The templates are available for online access, although they necessitate the use of FDA account credentials for the actual submission process.
The FDA Electronic Submission /FDA eSubmission
What is the FDA eSTAR?
The eSTAR is an interactive PDF template designed to facilitate the preparation of a comprehensive medical device pre-market submission for the FDA’s 510(k) clearance process in the US. Also, applicants can utilize the eSTAR for submitting responses to the FDA’s requests for additional information. Its aim is to improve the quality of submissions for various medical devices by ensuring that submitters provide complete and high-quality data for the FDA’s pre-market review.
By adopting the eSTAR format, submitters can be confident in the completeness of their submissions, which will, in turn, enable the FDA to conduct pre-market reviews more efficiently and ensure timely access to safe and effective medical devices. The eSTAR template is available free of charge and can be voluntarily used by all medical device submitters for 510(k), De Novo, and Q-submissions to the FDA. There are two types of eSTAR templates – one for medical devices and the other for IVDs. On June 09, 2023, the FDA released a beta version of eSTAR (PreSTAR) for pre-submissions (a type of Q-submissions).
Characteristics and Limitations of eSTAR Submissions
While the eSTAR program offers an interactive PDF form designed to assist applicants in crafting thorough medical device and IVD submissions, it comes with both advantages and disadvantages. Applicants must understand these benefits and limitations, based on which they can make well-informed choices about integrating eSTAR into their submission process. In the table below, we shall explore the attributes and restrictions of the eSTAR program.

Considerations for Effective eSTAR Usage
An advantageous feature of the eSTAR template, which contributes to the optimization of the submission process, is its automated incorporation of pertinent regulations and recognized standards for citation. This not only accelerates the submission process by reducing manual data entry but also mitigates the possibility of human errors that might occur during the input of regulations and standards. Through guided construction for each submission section, the utilization of eSTAR in the submission process can be effortlessly streamlined.
- The FDA suggests Adobe Acrobat Pro or Foxit PDF Editor for editing eSTAR templates.
- eSTAR includes certain integrated forms, thus eliminating the need for separate completion. These encompass Form 3514 (the submission cover sheet) and Form 3881 (indications of use). Additionally, the truthful and accurate statement, which was previously required on a company’s letterhead, is now integrated within eSTAR.
- eSTAR supports various attachment formats beyond just PDFs, such as Excel spreadsheets and video (mp4) files.
- Files in macro-enabled and executable formats are not permitted.
- The total size of the eSTAR PDF file, along with its attachments, must not exceed 1GB, as files larger than 4GB will not be accepted.
- If your electronic files surpass the technical limits, you can send the digital submission to the CDRH Document Control Center (DCC) via mail.
Best Practices for Preparing eSTAR Submissions
Following some of the best practices for preparing eSTAR submissions can help applicants streamline the submission process and improve their chances of a successful outcome. Here are a few best practices:
- Adhere to the FDA Guidance: The FDA offers guidance to help applicants successfully utilize the eSTAR template. Following this guidance ensures that submissions align with the FDA’s requirements and expectations.
- Ensure Fullness: The eSTAR template steers applicants through essential submission information. Applicants must furnish all required details to minimize the chances of deficiencies or additional information requests.
- Maintain Consistency: The eSTAR template fosters uniformity in the content and structure of 510(k) submissions. Guided construction for each submission section helps extract information on a medical device.
- Prioritize Clarity: Furnish clear and concise details in eSTAR submissions to facilitate a streamlined review process. Such detailing and precision minimize delays and errors, also ensuring timely access to pre-market submissions for medical devices.
- Secure Precision: Automatic verification of the information in each section of eSTAR submissions helps mitigate review delays and potential deficiencies.
Significance of Color Coding in FDA eSTAR Submission
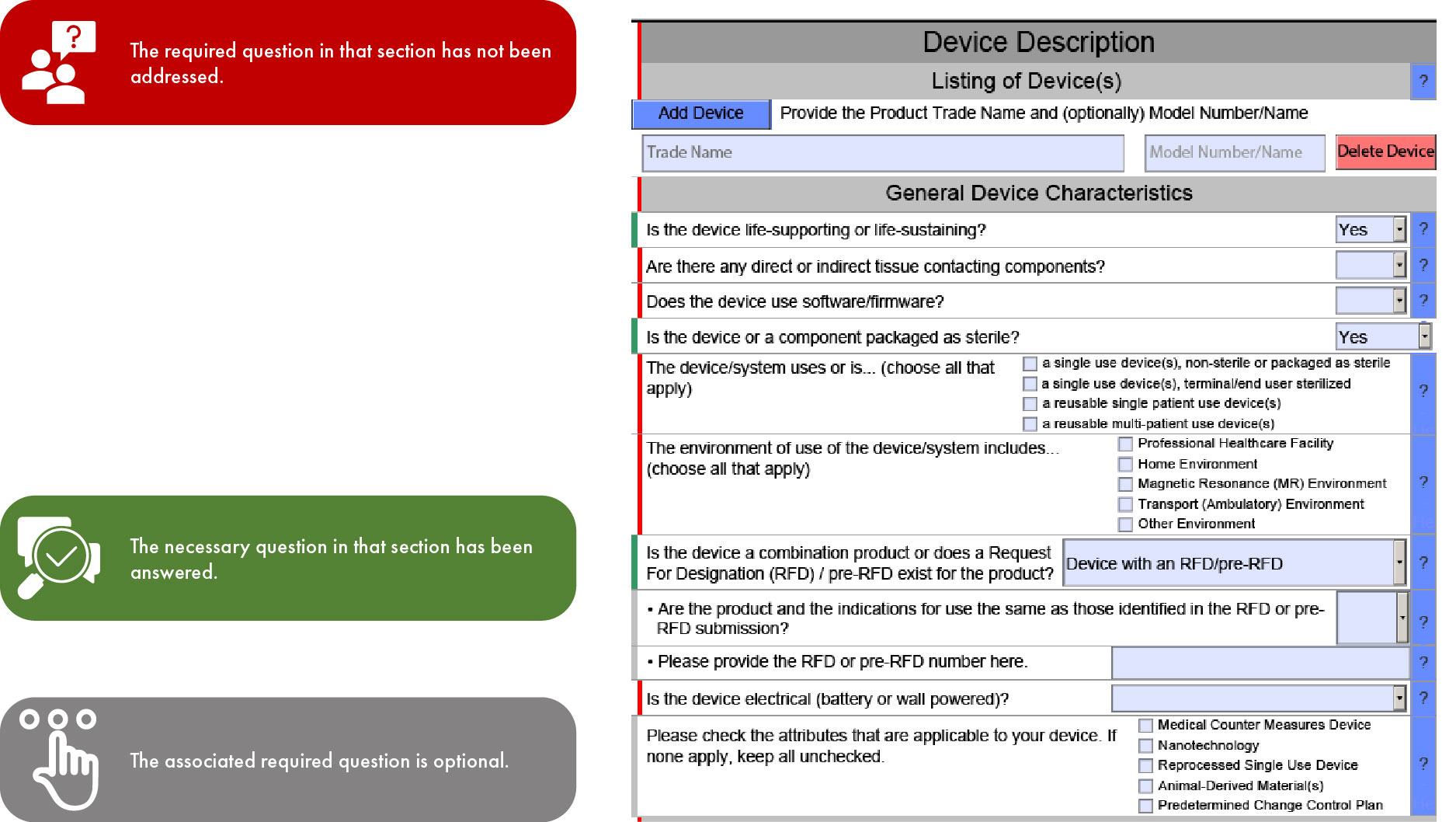
Application/Submission Types
Since eSTAR will become obligatory for 510(k) submissions starting October 01, 2023, and will be voluntary for De Novo, the template’s progression through sections is determined by the submission type. This kind of streamlining facilitates quicker review by the US FDA, thereby minimizing inconsistencies and omissions in your submission. However, it is important to note that the FDA might delay its review if English translations for the provided documentation are missing.

Align Your Attachments
Examine the eSTAR template beforehand to understand how attachments are divided and ensure that your documents align with the attachment sections. This step is crucial, particularly for companies with previous 510(k) submissions, as your existing approach toward document organization might require adjustments. It could involve extracting content from attachments as well.
- eSTAR demands concise attachments, which differ from traditional submissions.
- Expect numerous attachments, potentially dozens of them.
- Attachments allow the inclusion of data like Excel spreadsheets, MP4 video files, Word documents, JPEG files, etc.
- eSTAR wants page numbers for attachment information, but it does not suggest using a refusal-to-accept checklist for FDA guidance.
Templates: Templates are accessible in the provided Link: https://www.fda.gov/medical-devices/how-study-and-market-your-device/voluntary-estar-program.
The Future of eSTAR
It is anticipated that eSTAR will undergo updates upon the finalization of comprehensive applicable guidance documents. With these updates, the manufacturer can refer to the most recent versions issued by the US FDA
Health Canada (HC) has introduced a pilot initiative to employ eSTAR for Class III and IV devices. There exists a conjecture that other nations might emulate Canada’s lead in making the adoption of eSTAR discretionary, particularly within the IMDRF consortium. However, it should be noted that there have been no explicit declarations to this effect.
Why Choose Freyr?
Freyr boasts a wealth of expertise garnered from its involvement in numerous projects focused on submissions to the FDA in the past. By presenting dual pathways to proactively assist the industry in expediting 510(k) and De Novo submissions, Freyr provides an array of client services, which encompass a comprehensive list of essential submission documents and requisite information, conducting of meticulous gap analyses on documentation, compiling submissions via eSTAR, and finalizing the pre-market submission package to the FDA. Additionally, Freyr extends its support to post-submission activities, such as providing additional information and reviewing industry responses to any supplementary information requests posed by the FDA.
How Will Freyr Help?
Freyr offers support that may be helpful to get ready for 510(k) and De Novo submissions. The services include:
- Regulatory Pathway Support: This involves identifying the product code, regulation name and number, potential predicate/reference devices, performance testing, as well as the applicable standards and guidance documents pertinent to the specific device in question.
- Q-submission Support (Pre-submissions): This involves assisting you in clarifying queries related to the prerequisites of a pre-market submission, organizing the submission, getting ready for an FDA meeting, engaging with the FDA during the meeting in an interactive manner, and composing minutes of the meeting.
Freyr Expertise and Advantages
Expertise
Freyr Expertise
- Comprehensive FDA Regulatory strategy.
- Predicate device identification.
- Establishing substantial equivalence with predicate device.
- Gap Analysis for FDA compliance.
- Compilation of twenty-one (21) sections of the 510(k) technical file.
- Publishing and creation of the eCopy.
- Validation and submission of the eCopy.
- Liaising services for device approval.
- The addressal of RTA response and deficiencies.
- Consultation services for addressing deficiencies.
- Device Listing and FURLS database maintenance.