China Medical Device Registration Overview
China is one of the fastest-growing markets for Medical Devices, where the demand for devices is majorly met through imports. NMPA (National Medical Products Administration) ,formerly known as CFDA and the Centre for Medical Device Evaluation (CMDE) are responsible for the review of the Medical Device import registration applications of all the three (03) classes of devices (Class I, II & III). As per NMPA regulations, an NMPA legal agent must be appointed to serve as a representative of overseas pharmaceutical and medical device companies applying for China Medical Device Registration.
Regulatory Authority: NMPA (National Medical Products Administration) (formerly CFDA)
Regulation: State Council Order No.739
Authorized Representative: NMPA Legal Agent Required
QMS Requirement: YY/T0287-2017, ISO 13485:2016
Assessment of Technical Data: Centre for Medical Device Evaluation (CMDE)
Labeling Requirements: Decree No.6 of CFDA
Submission Format: eRPS
Language: English & Chinese
China Medical Device Classification
The device classification is defined in the National Medical Products Administration’s (NMPA’s) Medical Device Classification Catalogue (Announcement No. 104/2017), * or/and the rules in Order No. 15 for medical devices. The devices are classified into three (03) classes based on the risk criteria. Class I devices are low-risk devices and Class III devices are high-risk devices.
|
Device Class |
Risk |
|
I |
Low-Risk |
|
II |
Medium-Risk |
|
III |
High-Risk |
China NMPA Legal Agent Services
Foreign manufacturers without a physical office in China must appoint a China Authorized Representative (CAR) to market their devices in the country. The NMPA legal agent is entitled to manage registration and communicate with the NMPA before and after the registration. The certificate issued by the NMPA will appear on the registration certificate. The license, however, is owned by the manufacturer.
China Medical Device Registration
Class I devices undergo administrative review, whereas Class II and Class III devices undergo a thorough review process. The data and testing requirements vary based on the availability of predicates. Hence, Class II and Class III device manufacturers should also identify predicates to determine the clinical data requirements for their devices. The NMPA issues Record Filing and Registration Certificates for Class I and Class II/III devices, respectively.
Process flow
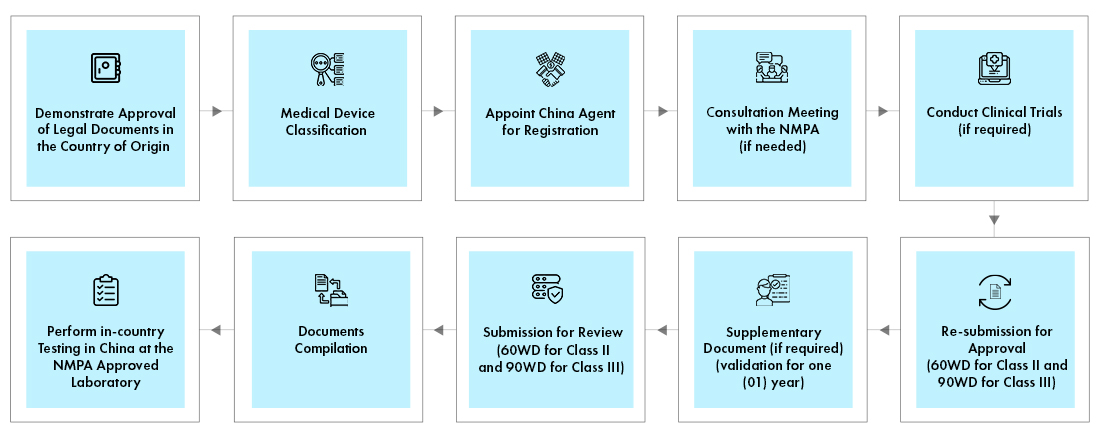
Post Approval Device Life Cycle Management
- Post approval change management - modifications to existing Medical Device approvals such as the addition of new variants, accessories; addition of new indications of use, among others
- Maintenance of approvals and registration through timely payment of administrative and registration fees
- Renewal of licenses
- Liaising between the NMPA and the manufacturer
- Importation management
With strong knowledge of the Chinese Regulatory landscape, Freyr guides manufacturers in successful clinical testing and the study data compilation as regulated by the NMPA. Freyr offers support in obtaining various certifications and compiling other documentation that leads to market approval within strict timelines. Freyr provides end-to-end Regulatory services for pre-and post-marketing activities of Medical Devices.
Summary
| Class | Registration Pathway | Review | NMPA Timelines | Validity of Registration |
|
Class I |
Notification |
Administrative Review |
One (01) day |
Unlimited |
|
Class II |
Registration |
Full application review |
1 – 2 Years |
Five (05) Years |
|
Class III |
Registration |
Full application review |
More than two (02) Years |
Five (05) Years |
Expertise
Freyr Expertise
- Regulatory due diligence for device registration with the NMPA
- Regulatory Intelligence (RI)
- Product license
- Production license
- In-country testing, including non-clinical and clinical testing
- Clinical Evaluation Report (CER)
- QMS compliance
- Compilation of device documents and GRP documents
- Labeling services
- Translation services
- Submission and Liaising services with the NMPA
- In-country representation
- Distributor identification and qualification