India CDSCO Medical Device Registration Overview
India is counted as one of the top global Medical Device markets with its major contribution from device imports. The Central Drug Standard Control Organization (CDSCO) regulates medical devices and In-Vitro Diagnostics (IVD) marketed in India. The CDSCO is headed by Drug Controller General of India (DCGI) and the approval authority is shared between Center Licensing Authority (CLA) and the State Licensing Authority (SLA). India CDSCO Medical Device Registration requires abiding by multiple regulations pertaining to device classification and patient safety standards.
Regulatory Authority: Central Drug Standard Control Organization (CDSCO)
Regulation: Medical Device Rules, 2017
Regulatory Pathway: Device Listing or Registration
Authorized Representative: Indian Authorized Agent (IAA) required for both Notified and Non-notified devices
QMS Requirement: Schedule 5 of MDR 2017/ ISO 13485:2016
Assessment of Technical Data: CDSCO or Notified bodies accredited by CDSCO
Validity of License: Unlimited
Labeling Requirements: Chapter VI of MDR 17, Legal Metrology Act
Submission Format: Online
Language: English
The Indian Medical Device Regulations are dynamic and are being made more stringent day by day. There are 37 categories of devices that are currently notified and regulated by the CDSCO in India. More device categories are being brought into purview of the CDSCO regulation in coming years. The Regulatory landscape in India is depicted as below:
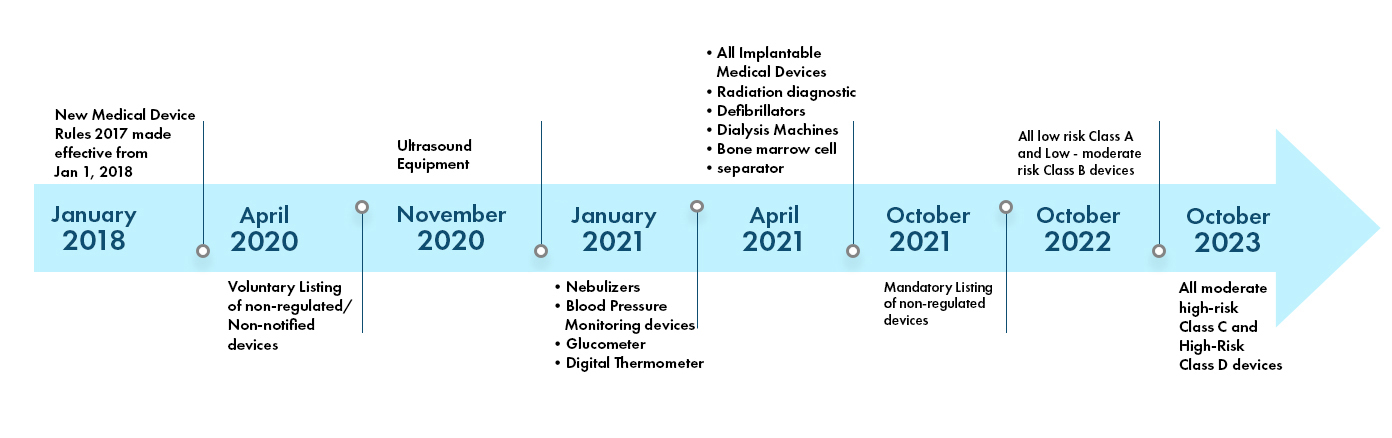
India Medical Device Classification
Separate classification systems exist for Medical Devices and Invitro Diagnostic Devices (IVD). Each of these categories are classified into 4 classes based on the extent of risk associated with these devices.
|
Device Class |
Risk |
|
A |
Low Risk |
|
B |
Low Moderate Risk |
|
C |
Moderate – High Risk |
|
D |
High Risk |
Indian Authorized Agent (IAA)
Foreign manufacturers should appoint an Indian Authorized Agent (IAA) to market devices in India. The IAA shall possess active wholesale drug license in the 20B & 21B application forms. Foreign manufacturers may appoint their distributors or importers as the IAA. However, having an independent IAA, with no commercial interest, would provide required flexibility to appoint multiple distributors in India. Click here to know more about the most frequently asked question on appointing an IAA.
India Medical Device Registration
All the applications for both Notified and Non-notified devices must be submitted through online portal called SUGAM and the CDSCO manages the applications through the same portal.
Non-Notified Devices: The devices not included in the list of Notified Devices are not regulated in India. However, these devices shall be listed in the CDSCO portal. Foreign manufacturers shall have an IAA based in India to market these devices.
Notified Devices: There are 37 categories of devices listed in Notified list, which are regulated by the CDSCO and require prior approval from the CDSCO for marketing them in India. There are different types of application forms and the documentation requirements vary with the application form submitted to the CDSCO.
The choice of application varies based on the home country approval, availability of Free Sale Certificate (FSC), nature of applicant, operations, and the type of device. After detailed evaluation of applications, the CDSCO issues licenses for either testing, import or for manufacturing of devices.
|
Intended Operation |
Indian Manufacturer (own Mfg. Site) |
Indian Manufacturer (Others Site) |
Foreign Manufacturer |
|
Device manufacturing for distribution or sales |
Manufacturing License |
Loan License |
NA |
|
Device manufacturing/ Importation for Testing purpose |
Test License |
Test License |
Test License |
|
Device Importation for distribution in India (with reference country approval) |
NA |
NA |
Import License without SEC Technical Assessment |
|
Device Importation for distribution in India (without reference country approval / FSC) |
NA |
NA |
Import License with SEC Technical Assessment |
Regulatory Approval Process Overview
Class A NSNM
- Appoint an importer to manage your registration and device importation in India. The importer must a valid wholesale license (Form 20B/21B or Form 41) or provide a valid justification if they lack a license
- Importer must have an online account on CDSCO portal
- Self declaration
- Mandatory registration of the product on the CDSCO portal
- Market the approved devices and IVDs in India
Class A
- Appoint an India Authorized Agent to represent you in interactions with the CDSCO. To manage your registration and device importation in India, The Authorized Agent must hold a valid wholesale license (Forms 20B and 21B/21C) and be granted Power of Attorney.
- IAA must have an online account on CDSCO portal
- For devices from non-reference countries, Class B license requires safety and performance verification through published data/clinical investigation and a free sale certificate from the country of origin
- Compilation of the device application (Form MD-14), including and not limited to manufacturing facility information, device technical information, ISO 13485 certificate, and IFU
- The registration/import license application must be lodged with CDSCO collectively with the relevant fees and documentation in English.
-
- The CDSCO evaluates the application and may request a technical presentation.
- A Subject Expert Committee (SEC) evaluates the Novel devices
- The CDSCO awards an Import License with no expiration date via Form MD-15. On the other hand, License retention charges are due every five years.
- Once authorized by CDSCO, the appointed India Authorized Agent (IAA) may import products.
- Market the approved devices and IVDs in India
Class B
- Appoint an India Authorized Agent to represent you in interactions with the CDSCO. To manage your registration and device importation in India, The Authorized Agent must hold a valid wholesale license (Forms 20B and 21B/21C) and be granted Power of Attorney.
- IAA must have an online account on CDSCO portal
- For devices from non-reference countries, Class B license requires safety and performance verification through published data/clinical investigation and a free sale certificate from the country of origin
- Compilation of the device application (Form MD-14), including and not limited to manufacturing facility information, device technical information, ISO 13485 certificate, and IFU
- The registration/import license application must be lodged with CDSCO collectively with the relevant fees and documentation in English.
-
- The CDSCO evaluates the application and may request a technical presentation.
- A Subject Expert Committee (SEC) evaluates the Novel devices
- The CDSCO awards an Import License with no expiration date via Form MD-15. On the other hand, License retention charges are due every five years.
- Once authorized by CDSCO, the appointed India Authorized Agent (IAA) may import products.
- Market the approved devices and IVDs in India
Class C
- Appoint an India Authorized Agent to represent you in interactions with the CDSCO. To manage your registration and device importation in India, The Authorized Agent must hold a valid wholesale license (Forms 20B and 21B/21C) and be granted Power of Attorney.
- IAA must have an online account on CDSCO portal
- Class C and IVD require in-country testing (clinical investigation/ clinical evaluation)through the National Institute of Biologicals (NIB) or a certified Indian lab or approved CRO(Clinical Research Organization) if they have no reference country approval
- Compilation of the device application (Form MD-14), including and not limited to manufacturing facility information, device technical information, ISO 13485 certificate, and IFU
- The registration/import license application must be lodged with CDSCO collectively with the relevant fees and documentation in English.
-
- The CDSCO evaluates the application and may request a technical presentation.
- A Subject Expert Committee (SEC) evaluates the Novel devices
- The CDSCO awards an Import License with no expiration date via Form MD-15. On the other hand, License retention charges are due every five years.
- Once authorized by CDSCO, the appointed India Authorized Agent (IAA) may import products.
- Market the approved devices and IVDs in India
Class D
- Appoint an India Authorized Agent to represent you in interactions with the CDSCO. To manage your registration and device importation in India, The Authorized Agent must hold a valid wholesale license (Forms 20B and 21B/21C) and be granted Power of Attorney.
- IAA must have an online account on CDSCO portal
- Class D and IVD require in-country testing (clinical investigation/ clinical evaluation)through the National Institute of Biologicals (NIB) or a certified Indian lab or approved CRO(Clinical Research Organization) if they have no reference country approval
- Compilation of the device application (Form MD-14), including and not limited to manufacturing facility information, device technical information, ISO 13485 certificate, and IFU
- The registration/import license application must be lodged with CDSCO collectively with the relevant fees and documentation in English.
-
- The CDSCO evaluates the application and may request a technical presentation.
- A Subject Expert Committee (SEC) evaluates the Novel devices
- The CDSCO awards an Import License with no expiration date via Form MD-15.on the other hand, License retention charges are due every five years.
- Once authorized by CDSCO, The appointed India Authorized Agent may import products.
- Market the approved devices and IVDs in India
Post Approval Device Life Cycle Management
Freyr supports the foreign manufacturers in end-to-end Medical Device lifecycle management, including post approval activities, such as:
- Post approval change management - modifications to existing Medical Device approvals such as, addition of new variants, accessories; addition of new indications of use among others
- Maintenance of approvals and registration through timely payment of administrative and registration fees
- Renewal of licenses
- Liaising between the CDSCO and the manufacturer
- Importation management
With a dedicated global delivery and operational centre in India, Freyr offers extensive knowledge on Indian Regulatory landscape. With a well-paved Regulatory strategy, Freyr provides end-to-end Regulatory services for pre- and post-marketing activities of Medical Devices.
Summary
|
Type of Device |
Risk |
Device Class |
QMS Audit |
Regulatory Pathway |
IAA |
Final Outcome |
|
Non-Notified Device |
NA |
NA |
NA |
Listing of devices in SUGAM portal |
Yes |
Listing in database |
|
Notified Device |
Low Risk |
A |
Schedule 5 of MDR 2017 or ISO 13485:2016 certification |
Device Registration |
Yes |
Test / Manufacturing /Loan/Import License |
|
Low - Moderate Risk |
B |
|||||
|
Moderate – High Risk |
C |
|||||
|
High Risk |
D |
DID YOU KNOW?
- CDSCO extends timeline for licensing of nebulizers, BP monitoring devices, digital thermometers, and glucometers
- Click here to view the CDSCO update
- 30th June, 2021 is the deadline
Submit your Application now!
Resource Center
Blogs
Case Studies
Infographic
What is...?
Expertise
Freyr Expertise
- Regulatory Intelligence and Due-Diligence services in India
- Medical Device Classification and grouping strategy in India
- Indian Authorized Agent (IAA) representation
- SUGAM account creation and account management
- Test License for Medical Devices and IVDs
- Import License for Regulated Medical Devices and IVDs
- Manufacturing License for Regulated Medical Devices and IVDs
- Loan license for Regulated Medical Devices and IVDs
- Listing of Non-Regulated Medical Devices
- Manufacturing Site Registration
- Compilation services for device documents and Device Master File (DMF)
- Liaison with the CDSCO, Central, and State Licensing Authorities
- In-country Testing services
- Post-Market Surveillance
- AERB approval for Radiation Diagnostic Devices