Mexico Medical Device Registration Overview
Mexico is the import hub for Medical Devices in Latin America. Mexico Medical Device Registration is regulated by the Federal Commission for the Protection against Sanitary Risk (Comisión Federal para la Protección contra Riesgos Sanitarios, COFEPRIS). Foreign manufacturers must appoint a Mexico Registration Holder (MRH) for registering their device.
- Diagnostic Agents
- Medical Equipment (I.E. Accessories And Appliances)
- Prosthetics, Orthotics, And Functional Aids
- Surgical Materials
- Hygiene Devices
- Dental Supplies
Regulatory Authority: COFEPRIS
Regulation: Pharmaceutical Affairs Act (PAA) & Regulation for registration of Medical Devices
Regulatory Pathway: Device Registration
Authorized Representative: Mexico Registration Holder (MRH)
QMS Requirement: Quality System Documentation (QSD) ISO 13485
Assessment of Technical Data: COFEPRIS
Validity of License: 5 years
Submission Format: Paper
Language: English & Spanish
Mexico Medical Device Classification
COFEPRIS classifies devices into Class I, II and III based on 20 rules framed by Health Products Technical Committee. The rules intended for Medical Device Regulation in Mexico are very broad needing extensive understanding. The basis for classification is ambiguous to decode and the challenges pertaining to linguistic translations are also prevalent. After device are placed in their groups, they are categorized into four different classes depending on their risk: Low Risk, Class I, Class II, Class III. Class I Medical Devices are associated with the lowest risk, while Class III Medical Devices are associated with the highest risk.
Mexican Registration Holder (MRH)
Foreign manufacturers should appoint a Mexican Registration Holder (MRH) as a pre-requisite to market devices in Mexico. Foreign manufacturers may appoint their distributors or importers as SRP. The SRP shall, however, provide required flexibility to appoint multiple distributors in Mexico.
Mexico Medical Device Registration
The manufacturers may opt for any of the three pathways for device registration - Traditional pathway, Equivalence pathway, and/or Third-Party review of devices.
Class I devices do not require to undergo detailed device review process and a simple notification would suffice to market the device in Mexico.
Class II and Class III Devices with approval in reference countries such as, the U.S., Canada or Japan qualify for Equivalency Review process. Devices without these approvals shall undergo standard Review process. Applicants may opt to go through the Third-Party Review (TPR), which is faster at the cost of additional expenses payable to such accredited Third-Party.
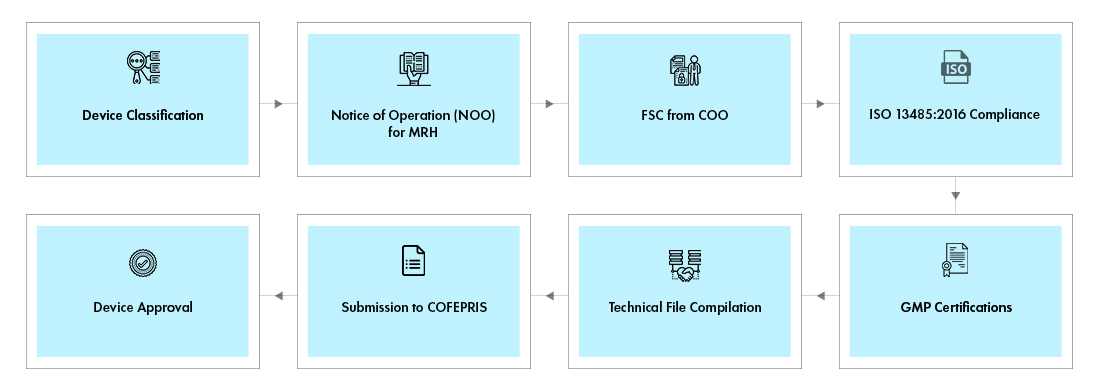
Process flow
Post Approval Device Life Cycle Management
Freyr supports the foreign manufacturers in end-to-end Medical Device lifecycle management, including post approval activities, such as:
- Post approval change management - modifications to existing medical device approvals such as, addition of new variants, accessories; addition of new indications of use among others
- Maintenance of approvals and registration through timely payment of administrative and registration fees
- Renewal of licenses
- Liaising between the COFEPRIS and the manufacturer
Summary
|
Class |
Registration Pathway |
Timelines |
|
I |
QMS + Product Registration |
1-3 months |
|
II |
QMS + Product Registration |
10 months - 1 years |
|
III |
QMS + Product Registration |
10 months - 1 years |
Expertise
Freyr Expertise
- Regulatory Due Diligence
- Official Classification
- Notice of Operation support
- Device Registration
- Mexican Registration Holder (MRH)
- Labeling support
- Translation support
- Distributor identification and qualification
- Post Marketing Surveillance
- Post Approval Change Management
- License renewal and transfer
- Submission and liaison with the COFEPRIS
Freyr Advantages