Europe Medical Device Registration Overview
The EU consisting of 27 member states developed the Medical Devices Regulation (MDR) 2017/745 and In Vitro Diagnostics Medical Devices Regulation (IVDR) 2017/746 which were wholly implemented recently. These regulations are an integral part of Europe medical device registration process and have now replaced the Directives. Both the regulations consist of new additional requirements, and these will be the centralized regulation procedures that will be required to follow in placing the medical devices in any of the 27 countries. Foreign Medical Device manufacturers who do not have a physical location in Europe must appoint a European Authorized Representative (EAR) to help them comply with these regulations.
Regulatory Authority: European Medicines Agency (EMA)
Regulation: Medical Device Regulations (MDR) 2017/745, In Vitro Diagnostic Devices Regulations 2017/746
Regulatory Pathway: CE marking followed by Registration/Notification
Authorized Representative: European Authorized Representative (EAR) for Non-EU manufacturers
QMS Requirement: ISO 13485:2016
Assessment of Technical Data: Notified Body for CE marking
Validity of License: Unlimited
Submission Format: Paper
Device Classification
Classification of the device is the very first step in determining the regulatory pathway for the given product. There are about 22 implementing rules for medical devices and are classified as –
|
Class |
Risk |
|
Class I |
Low |
|
Class IIa |
Moderate |
|
Class IIb |
Moderate to High |
|
Class III |
High |
Similarly, for IVD, about 7 rules are implemented while classifying into the following four categories
|
Class |
Risk |
|
Class A |
Low |
|
Class B |
Moderate |
|
Class C |
Moderate to High |
|
Class D |
High |
Given the specialized instructions in place for different classes, identifying the right class of device is crucial in determining the Regulatory pathway.
European Authorised Representative (EAR)
Any foreign manufacturer intending to launch their devices in the EU region is obligated to appoint a European Authorised Representative (EAR) in accordance with Article 11 of the EU MDR and IVDR.
Medical Device Registration
In order to market the medical devices in the EU geography, obtaining CE marking is mandatory. Manufacturers are required to identify and appoint notified, go under the conformity assessment, and issue CE certification.
Decoding Regulatory information pertaining to device registration or notification via an online registration system can prove challenging without an expert’s assistance.
Process flow
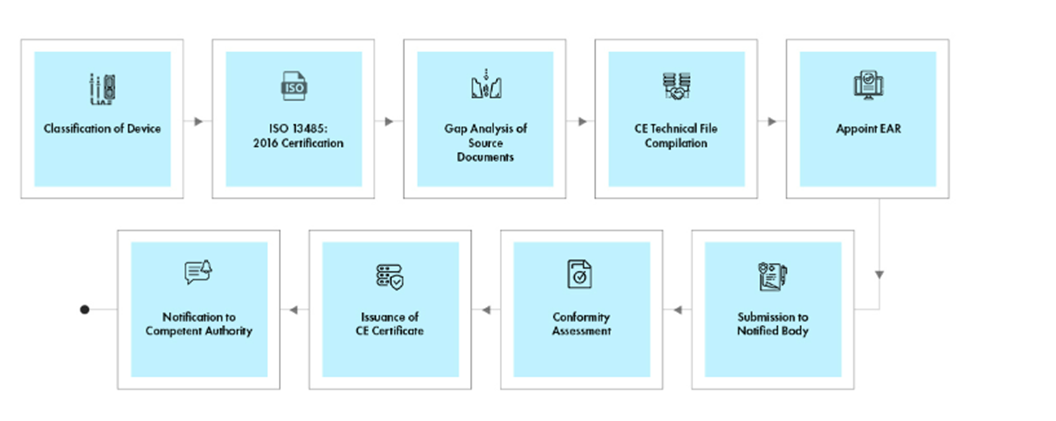
Post-approval Device Lifecycle Management
The European medical devices regulations highlight now on the importance of post-market requirements. The manufacturer is required to be equipped with a surveillance management system. A periodic information is to be provided for the device.
Freyr can support you in building post-market surveillance (PMS) plan, the post-market surveillance report (PMSR), periodic safety update report (PSUR), and post-market clinical follow-up (PMCF)/ post-market performance follow-up (PMPF).
Freyr supports also includes activities such as –
- Post approval change management - modifications to existing medical device approvals such as, addition of new variants, accessories; addition of new indications of use among others
- Maintenance of ISO 13485:2016 and CE certification
- Renewal of licenses
- Liaising between Notified body and the manufacturer
Expertise
Freyr Expertise
- European Medical Device Classification
- European Authorized Representative (EAR) support
- ISO 14971:2019 Risk Management consultation
- ISO 13485:2016 Compliance
- CE technical file/design dossier review, compilation and submission
- EU MDR Transition Support
- EU IVDR Transition Support
- Clinical Evaluation Reports (CER) for Medical Devices
- Performance Evaluation Reports (PER) for In Vitro Diagnostic Devices
- Notification/Registration of Medical Devices via Online Registration System
- Medical Device Regulatory strategy report
- Testing support- biocompatibility, electrical safety, mechanical and performance
- Labeling Compliance support
- GMP support
- Post-market Surveillance support