Overview
Aperçu des services de mise en conformité MDR de l’UE
Le règlement de l'UE sur les dispositifs médicaux (MDR) est entré en vigueur le 26 mai 2021, après un délai de transition de trois ans et une prolongation d'un an en raison de la pandémie de COVID-19. Les dispositifs mis sur le marché de l'UE doivent désormais être conformes à ces réglementations et doivent être certifiés CE conformément au règlement MDR de l'UE par les organismes notifiés accrédités dans le cadre de ces réglementations. Les dispositifs qui ont déjà été certifiés CE conformément à la directive MDD de l'UE bénéficient toutefois de délais de grâce avant de devoir se conformer pleinement aux exigences MDR de l'UE. Pendant cette période de grâce, les dispositifs certifiés à la fois selon la MDD et la MDR de l'UE coexisteront sur le marché avec le même statut et sans faire l'objet d'une discrimination. Freyr offre des services inégalés de mise en conformité MDR de l'UE pour aider les entreprises de dispositifs médicaux à répondre aux exigences MDR de l'UE dans les délais impartis.
Prendre rendez-vous avec nos experts MDR de l'UE
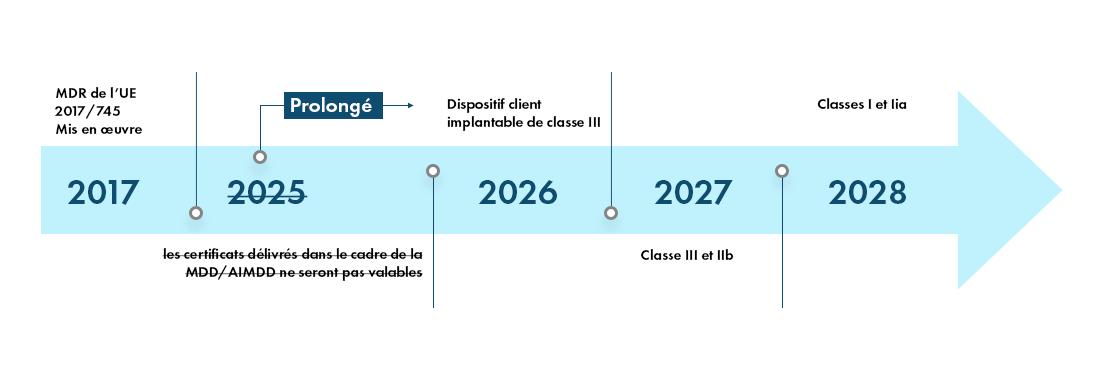
Le MDR - Calendrier de transition et nouvelles classifications des dispositifs
Le règlement européen sur les dispositifs médicaux (MDR) entrera pleinement en vigueur dans tous les États membres de l'UE et les États membres de l'association européenne de libre-échange (EFTA) à partir de mai 2021 et offre aux fabricants une période de transition de quatre ans pour obtenir la certification MDR de l'UE.
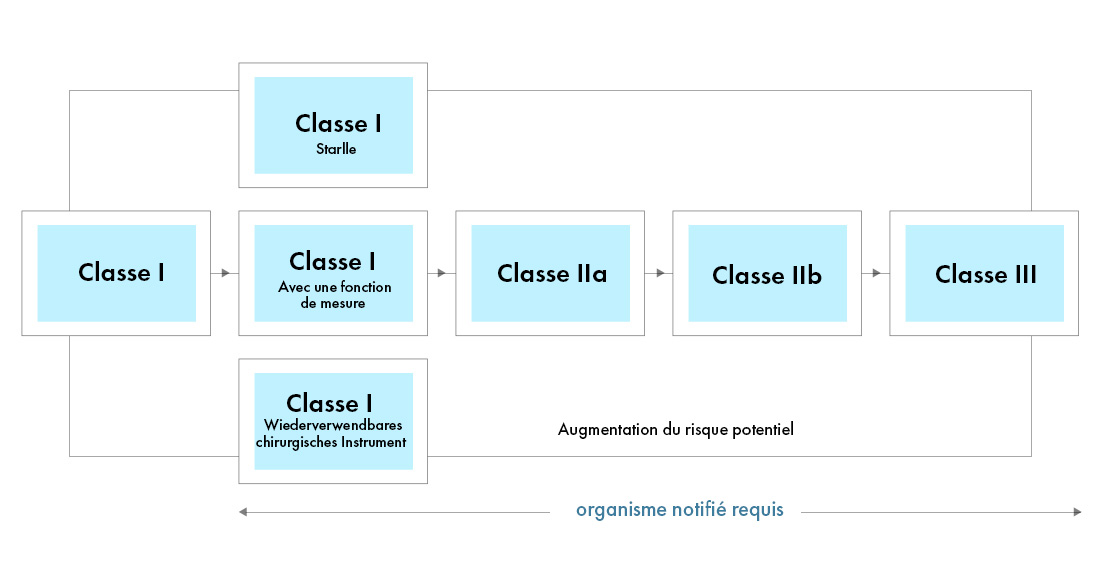
Le nouveau règlement européen sur les dispositifs médicaux (MDR), tel qu'il a été observé, a également apporté des modifications au système de classification des dispositifs existant, notamment :
De l'identification des changements exacts à apporter à leur mise en œuvre en temps réel, les fabricants peuvent être amenés à relever toute un ensemble de défis pour se conformer aux exigences du règlement MDR de l'UE. Qu'il s'agisse du décodage de la nouvelle structure, de la classification précise d'un dispositif, de la collecte et de la soumission de toutes les données, les fabricants devront adopter une approche réglementaire plus détaillée et plus transversale pour se conformer à la nouvelle réglementation européenne sur les dispositifs médicaux. Grâce à une analyse rigoureuse des lacunes, Freyr aide ses clients à maintenir le statu quo et à prendre les mesures réglementaires nécessaires à la transition et à la mise en conformité avec la MDR de l'UE.
Obtenez des conseils d'experts sur votre mise en conformité avec la MDR de l'UE
Freyr Expertise and Advantages
L'EXPERTISE
L'EXPERTISE FREYR
- Développer une stratégie claire de mise en œuvre du règlement sur les dispositifs médicaux (MDR)
- Comprendre la nouvelle législation, effectuer une analyse des lacunes par rapport aux systèmes de gestion de la qualité (SGQ) actuels et aux processus en place.
- Élaboration d'un plan détaillé avec une approche interfonctionnelle pour déterminer les aspects du système de qualité qui devront être modifiés conformément au nouveau règlement de l'UE sur les dispositifs médicaux.
- Former plusieurs équipes pour analyser le champ d'application du produit, la classification, le traitement du système de gestion de la qualité, etc. au sein de l'organisation, avec un point de contact unique dans chaque équipe.
- Affectation et planification des ressources
- Prendre en compte l'interaction de votre SGQ avec d'autres règlements et utiliser cette opportunité pour rationaliser les processus, tout en permettant la flexibilité nécessaire pour intégrer de futurs changements.
- Analyser les données de test en place et vérifier s'il existe des exigences supplémentaires imposées par le MDR.
- Coordonner les attentes et le plan de transition avec les organismes notifiés de l'UE
- Analyse des lacunes des dispositifs médicaux existants entre les règlements MDD et MDR de l'UE
- Soutien de bout en bout à l'élaboration du rapport d'évaluation clinique (CER), y compris la recherche documentaire conformément aux lignes directrices du règlement européen sur les dispositifs médicaux (EU MDR).
- Services de bout en bout pour les rapports de surveillance post-marché (PMSR), les rapports périodiques de sécurité (PSUR) et les résumés de sécurité et de performance clinique (SSCP).
- Augmentation des ressources réglementaires avec des options de déploiement onshore et offshore
- Services du représentant autorisé de l'Union européenne (EAR)
- Conformité au règlement MDR et assistance à la transmission des données aux organismes notifiés
- Veille réglementaire couvrant le processus d'importation des différents marchés réglementés
- Conformité du SMQ et simulations d'audits
- Système de gestion des documents et outil pour les entreprises MDR
- Classification et reclassification des dispositifs en fonction du risque
- Mise en œuvre et consultation de l'UDI
- Services de surveillance post-marché conformes au règlement de l'UE sur les dispositifs médicaux
- Gestion des risques ISO 14971 consulting
- Formation interne et en ligne
- Personne responsable des services de conformité à la réglementation et de l'assistance
- Identification des organismes notifiés MDR
Pour une assistance réglementaire de bout en bout sur le MDR de l'UE, contactez Freyr